Bachelorstudium (Studienstart bis WS19/20)
Das Department Chemie und Pharmazie bietet zwei Bachelorstudiengänge an
Ab dem WS 2022/23 werden alle Veranstaltungen des Bachelorstudiums nach der Prüfungsordnung 2020 (PO2020) angeboten. Studierende nach der PO2013 (Studienstart WS 19/20 oder früher) wenden sich für Fragen zum weiteren Studienverlauf und/oder zur Belegung von Veranstaltungen bitte an Prof. Burzlaff.
Informationen zum Stundenplan, Prüfungsterminen etc. finden Sie unter ‚Bachelorstudium‘.
Übersicht über das Bachelorstudium
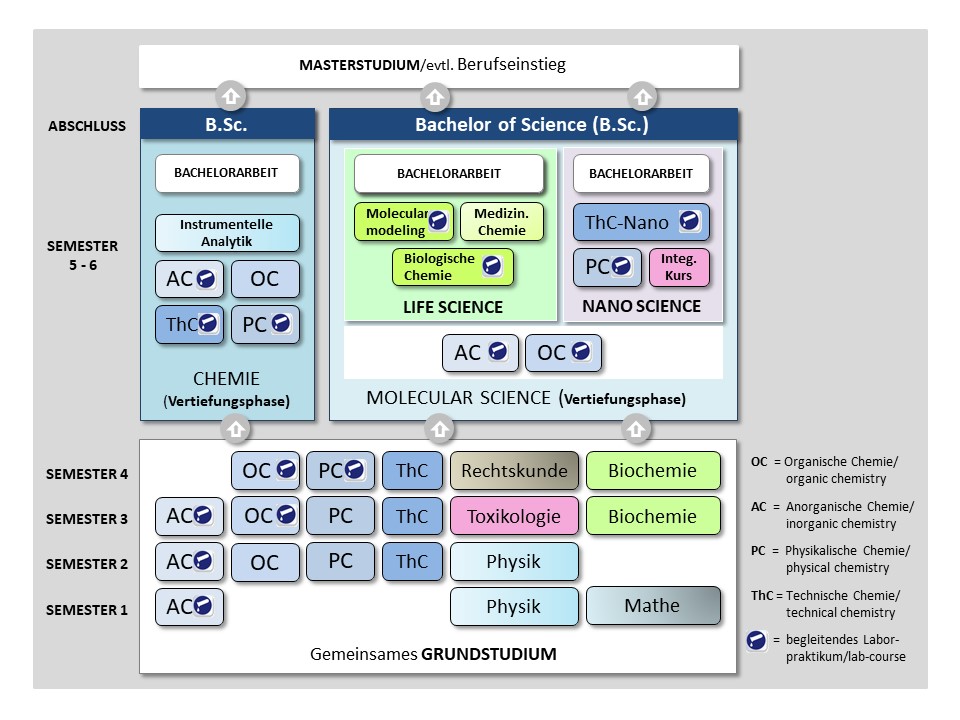
Die Studieninhalte sind über die Prüfungsordnung festgelegt und im Modulhandbuch BSc Chemie bzw. Modulhandbuch BSc Molecular Science näher ausgeführt.
Das Bachelorstudium ist in zwei Studienphasen gegliedert
Im Grundlagenstudium (1.-4. Semester) gehören neben den Grundlagen in den klassischen chemischen Disziplinen Anorganische und Allgemeine Chemie, Organische Chemie, Physikalische Chemie sowie Theoretischen Chemie weitere Grundlagenfächer wie Mathematik, Physik, Biochemie und Molekularbiologie, Toxikologie und Rechtskunde zum Lehrplan.
In der Vertiefungsphase (5./6. Semester) wird das Profil entsprechend des Studiengangs ausgebildet, der Abschluss zum Bachelor of Science erfolgt durch eine eigenständige wissenschaftliche Bachelorarbeit.
In Chemie erfolgt die Vertiefung durch den Ausbau der Kenntnisse aus dem Grundstudium. So werden vertiefte Einblicke in die Kernfächer der Chemie – Anorganische Chemie, Organische Chemie und Physikalische Chemie – erworben, hierbei wird in der Vertiefungsphase insbesondere Wert auf eine fundierte präparativ-experimentelle Ausbildung gelegt. Des weiteren werden die Kenntnisse in Theoretischer Chemie und Instrumenteller Analytik, hier mit Schwerpunkten in der Spektrometrie und Spektroskopie, intensiviert.
In Molecular Science erfolgt die Vertiefung entspechend der Wahlrichtung Molecular Life Science oder Molecular Nano Science. Neben der Vertiefungsrichtung erfolgt eine intensivierende Ausbildung in der Anorganischen und Organischen Chemie.
Im Bereich „Life Science“ wird auf der Basis der erworbenen Grundlagen den Studierenden ein fundierter Quereinstieg im Bereich Wirkmechanismen und Wirkstoffdesign von Arzneistoffen ermöglicht. Dies erfolgt durch die koordinierte Vermittlung der fachlichen Grundlagen aus den Bereichen Genetik, Biochemie, Mikrobiologie, molekularer Pflanzenphysiologie, Medizinische Chemie und Lebensmittelchemie. Ein wichtiger Aspekt ist hierbei die Einbindung moderner, computergestützter Struktursuche- und Strukturoptimierungsverfahren.
Im Bereich „Nano Science“ greift die Entwicklungen auf dem Gebiet neuer, molekularer Materialien im Bereich Werkstoff- und Materialwissenschaften, wie z.B. leitende organische Verbindungen oder anorganische und organische Solarzellen, auf. Hier werden die physikalischen und chemischen Eigenschaften, der Aufbau der Materie und deren Wechselwirkung mit Licht sowie verschiedene spektroskopischer und spektrometrischer Analysemethoden vermittelt.