„Ca“-talysis: Katalyse mit Kalzium
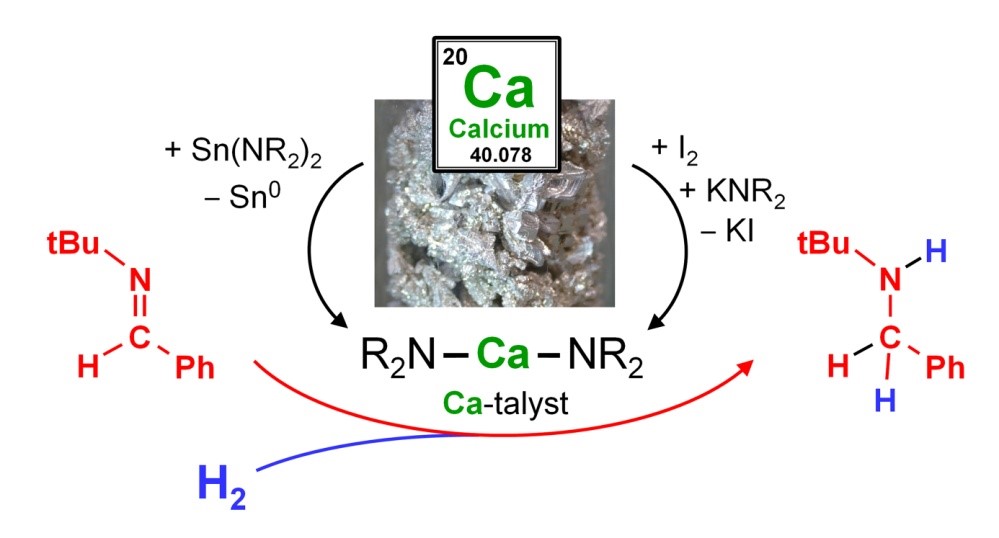
Katalyseforschung mit günstigen und biokompatiblen frühen Hauptgruppenelementen entwickelt sich derzeit atemberaubend schnell. Vor allem Calcium spielt eine Schlüsselrolle bei der Überschreitung von Grenzen, die bisher unangetastet blieben. Die Forschungsgruppe von Professor Sjoerd Harder, Lehrstuhl für Anorganische und Metallorganische Chemie am Department für Chemie und Pharmazie der FAU, leistet Pionierarbeit in diesem Gebiet. Sie hat es sich zur Aufgabe gemacht, das volle Anwendungspotential insbesondere der Erdalkalimetalle bei anspruchsvollen, modernen Katalyseprozessen zu demonstrieren: Einfache Hauptgruppenmetalle können in vielen Fällen die traditionell genutzten und etablierten Übergangsmetalle, so wie Platin oder Palladium, in der Katalyse ersetzen – ein Paradigmenwechsel in der Metallorganischen Chemie.
Ihr jüngster Beitrag wurde in der allerersten Ausgabe von Nature Catalysis publiziert. Das Journal ist ein neues Mitglied in der Gruppe der Nature Fachzeitschriften. Darin wird die Erdalkalimetall-katalysierte Hydrierung von Iminen mit H2 bei erstaunlich niedrigen Drücken (bis zu 1 bar) mit extrem simplen und leicht zugänglichen Katalysatoren präsentiert. Die Autoren beschreiben ihre überraschende Entdeckung als unerwartete katalytische Transformation. Die veröffentlichte Arbeit entstand in einer Kooperation mit der Universität Brüssel, wo theoretische Berechnungen zum Mechanismus der Reaktion angestellt wurden. Betrachtet man die Effizienz der Umsetzung, die breite Anwendbarkeit und die Toleranz funktioneller Gruppen, so bedeutet diese Arbeit einen weiteren Meilenstein in dem sich schnell entwickelnden Gebiet der Katalyse mit frühen Hauptgruppenmetallen.
Publikation
H. Bauer, M. Alonso, C. Färber, H. Elsen, J. Pahl, A. Causero, G. Ballmann, F. de Proft, S. Harder Nature Catalysis 2018, 1, 40-47.
Siehe auch die Rubrik News & Views in Nature: D. Stephan, Nature 2018, 553, 160-162.
Kontakt
Prof. Dr. Sjoerd Harder, PhD
91058 Erlangen
- Telefon: +49913185-27350
- E-Mail: sjoerd.harder@fau.de
- Webseite: https://www.chemie.nat.fau.de/person/sjoerd-harder/